From Single Motif to Active Ensembles: Phase-Controlled Co8 Cluster Catalysis on MoS2 for Nitrogen Electroreduction

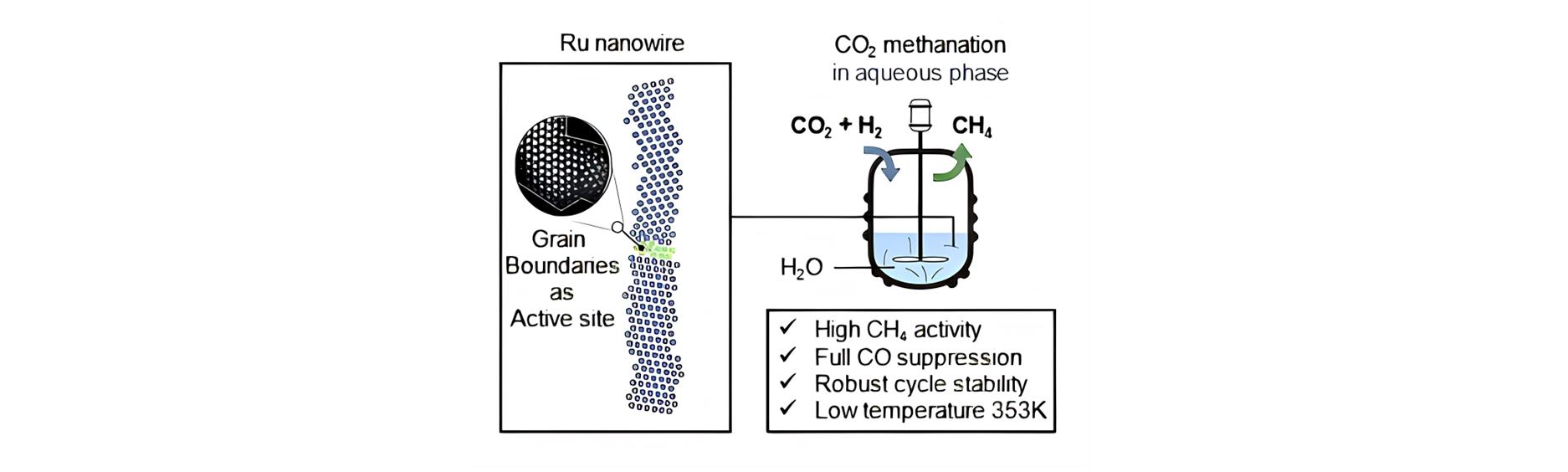
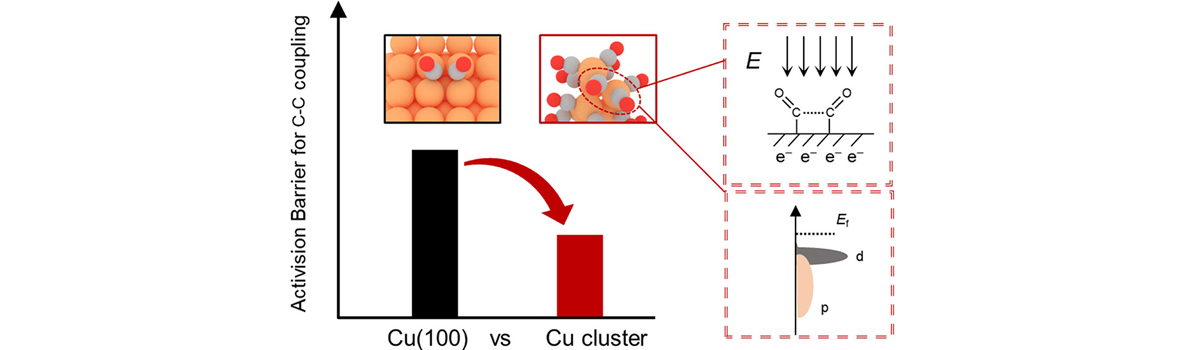
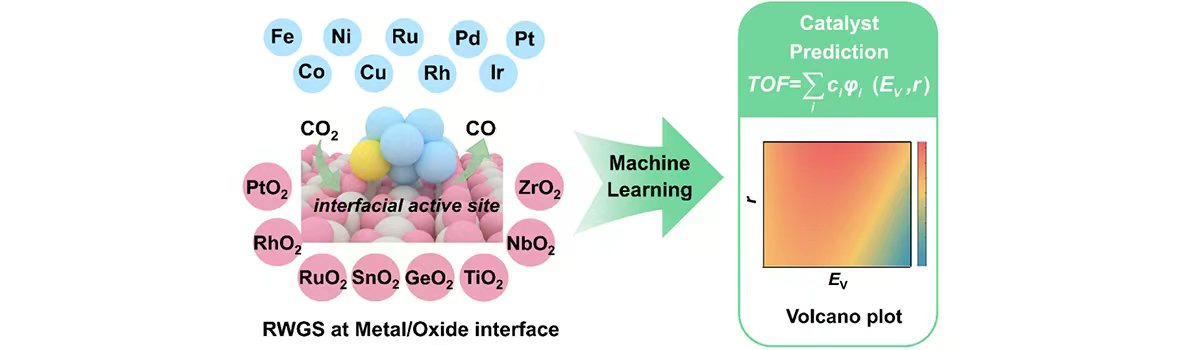
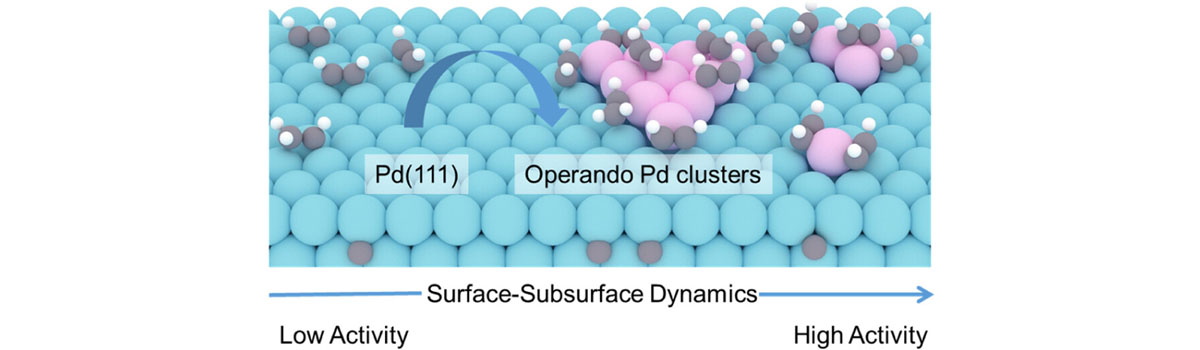
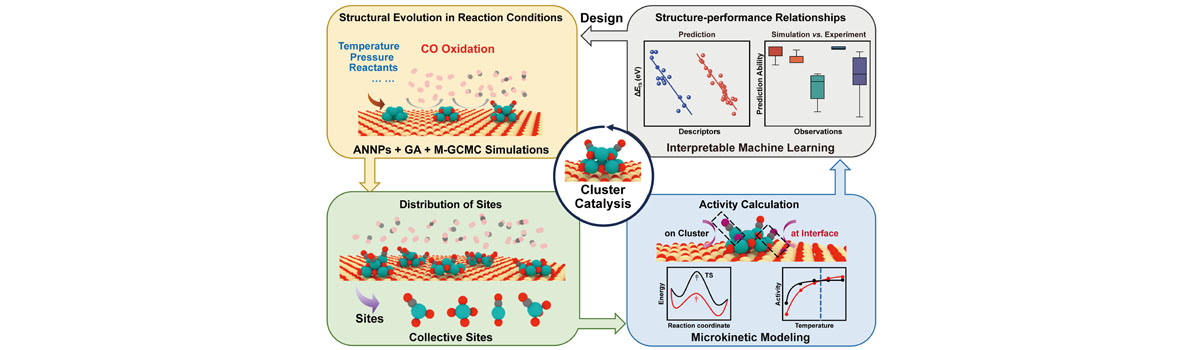
Our group has expertise in first-principles computational modeling and machine learning approaches to understand and predict catalysts for use in energy storage, sustainable chemical production and reducing the environmental impact of products. Developing and using new theoretical approaches, we devoted to establish the structure-activity relationship of the catalytic materials at atomic scale to realize the rational design of better catalysts. The interests of our group include: (1) Integrated Genetic Algorithm - Grand Canonical Monte Carlo - Microkinetics simulations approaches together to study the active structure of clusters catalysts, and to clarify the influence of the dynamic evolution of structures on catalytic performance; (2) The theoretical investigation of crystal phase effect on heterogeneous catalysis and electrocatalysis; (3) Using machine learning approach to accelerate catalyst and materials design. Our group has published more than 40 peer-reviewed papers, including Nat. Commun., Natl. Sci. Rev., J. Am. Chem. Soc., Angew. Chem. Int. Ed. and so on. We welcome students of all ages, backgrounds and beliefs to join our group.