The interests of our group focus on theoretical catalysis, including: structure sensitivity of nanocatalysts, reaction mechanisms, and nanoscale effects in heterogeneous catalysis and electrocatalysis using DFT, as well as molecular simulation, and machine learning. The main results of our recent works are described below:
1) Crystal phase effect in Fischer-Tropsch synthesis.
CO activation under hydrogen atmosphere condition as a probe reaction was considered in our work. We discovered that the Co crystalline phase plays a significant role in CO activation: (1) Hexagonal close packed (HCP) Co has higher intrinsic activity than face centered cubic (FCC) Co, and the catalytic activity is structure dependent, and (2) CO prefers direct dissociation on HCP Co catalyst, whereas CO prefers the hydrogen-assisted pathway on FCC Co. We found that HCP Co can expose highly active surfaces due to its low crystal symmetry. Based on the theoretical findings, a new catalyst structure with high mass specific activity by exposing highly active HCP Co (10-11) and (11-21) facets, without decreasing the catalyst size, was suggested. The great influence of crystallographic structure and corresponding morphology effect on the formation of the active sites with higher intrinsic activity and density was illustrated in this study. The insights revealed might open a new avenue for the design of better, stable catalysts with maximum mass-specific reactivity, in which ab-initio calculation and material synthesis would play an essential role. (J. Am. Chem. Soc., 2013, 135 (44), 16284–16287)
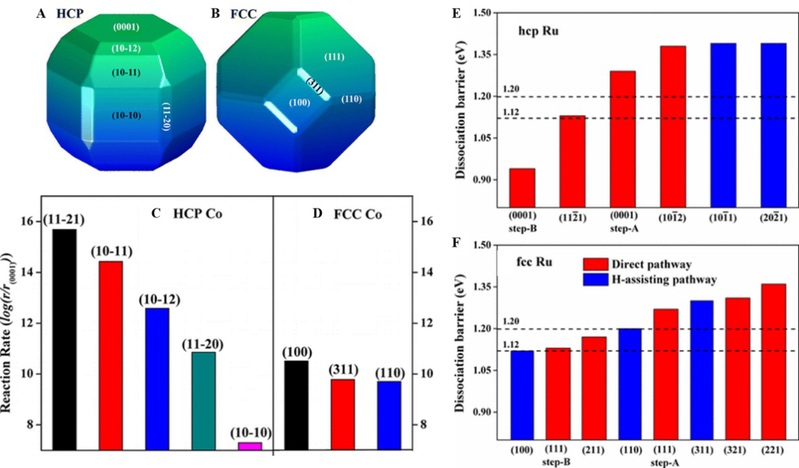
Crystal phase effect on CO activation in Fischer-Tropsch synthesis.
Inspired by Co work, we demonstrate a strategy for tuning the crystal phase of Ru catalysts to expose a high number of active sites for a higher mass-specific activity. Density functional theory calculations show that upon CO dissociation there are several open facets with modest barrier available on the FCC Ru but only a few step edges with a lower barrier on conventional HCP Ru. Guided by theoretical calculations, we have synthesized Ru catalysts with FCC phase. The mass-specific activity of the FCC Ru catalysts is much higher than the previous best HCP catalyst with a smaller size of 1.9 nm and a higher specific surface area. The origin of the higher mass-specific activity of the FCC Ru catalysts is identified experimentally from the 2 orders of magnitude higher density of the active sites, despite its slightly higher apparent barrier. Experimental results are in excellent agreement with theoretical predictions. The impact of the crystal phases on active site distribution and their intrinsic activities revealed here provides a route to rationally design catalysts with higher mass-specific activity (without decreasing the particle size). (J. Am. Chem. Soc., 2017, 139 (6), pp 2267–2276)
Ni has the opposite catalytic behavior as compared with Co. On both FCC and HCP Ni, CO dissociation shows a dramatic structural sensitivity irrespective of direct or H-assisted pathway. Compared to the direct dissociation, the H-assisted dissociation is kinetically favorable on both FCC and HCP Ni. With increasing the dissociation barrier, the preferred dissociation pathway changes from a COH intermediate to CHO intermediate. FCC Ni can expose abundant facets with low barrier compared with HCP Ni. Our work will help optimize more active Ni catalysts for the CO methanation reaction. (J. Phys. Chem. C, 2016, 120 (43), pp 24895–24903) In view of our excellent work in crystal phase effect investigation, we were invited to write the two review papers: (1) WIREs Comput Mol Sci 2016, 6:571–583 and (2) Engineering 3 (4), 467-476.
2) A novel computational strategy to identify the active phase structure in heterogeneous catalysts
The ceria-supported palladium (Pd/CeO2) catalyst has attracted widespread attention due to its excellent catalytic performance in combustion processes. The high catalytic performance of Pd/CeO2 is usually understood in terms of strong metal-support interactions (SMSI), which maintain a high Pd dispersion and can also affect the oxidation state of the Pd. An important property of ceria is its ability to reversibly store O atoms. Consequently, many investigations of low-temperature CO oxidation over Pd/CeO2 focus on the oxidation state of Pd, the role of ceria O vacancies, and the specific topological features of the Pd-CeO2 interface. There is an ongoing debate about the oxidic or metallic nature of the active phase in Pd/CeO2.
Selection of a suitable model for computational modeling of reaction mechanism is important but remains challenging for small supported clusters. For periodic surfaces or large nanoparticles, the ab initio thermodynamics approach introduced by Scheffler and Reuters can be used. In the present study, we developed alternative approaches to predict the shape of a Pd8 cluster supported on CeO2(111), at the DFT level using a genetic algorithm (GA-DFT) and to predict shape and composition of the supported Pd8 in contact with CeO2(111) using a basin-hopping-based approach in the Gibbs ensemble (GCMC-DFT). The GA-DFT approach shows that the Pd clusters grows into a structure commensurate with the ceria surface topology. The reactivity of the Pd-CeO2 interface is much higher than that of a gas-phase Pd8 cluster. In contact with oxygen, the Pd8 cluster is oxidized. Microkinetics simulations predict that the CO oxidation activity of the oxidized clusters is much higher than for the reduced clusters, which can be explained by the decreased CO binding energy upon oxidation of Pd8. This work presents an advanced approach for determining the active phase structure and composition under practical reaction conditions, which we expect to become a standard given the rapid advances in computational power. (J. Am. Chem. Soc., 2018, 140, 4580 (Journal Cover))

Integrated “GA-GCMC-DFT-MKM” together to identify the active structure under atmosphere.
The latest generation of supported catalysts has pushed the material utilization efficiency of expensive and often scarce metals to their upper limit as single atoms, where 100 % metal dispersion is obtained. In this single-atom limit, it is unfeasible to continue elevating the catalytic activity by improving the metal dispersion for a given catalytic formulation because the already isolated atoms cannot be further dispersed. Therefore, the current technical route often limits itself to increasing the number of anchoring sites on a support for such a single-atom species to develop catalysts that are often incrementally better. Alternatively, if an improved active site may have more than one metal atom, the question becomes how we can effectively build an optimized multi-atom ensemble site with an appreciable amount on a given catalyst support. To overcome the stalled catalytic efficiency set by single Pt atoms on ceria, we introduced a general approach of going beyond the current technical route of single-atom catalyst development—we use the single Pt atoms as “seeds” to assemble an orders of magnitude more active catalytic site while keeping 100 % Pt utilization. By GCMC simulations combined with microkinetic simulations, we found Pt cluster is more active than the single-atom catalysts. (Nat. Commun. 2019, 10, 3808)
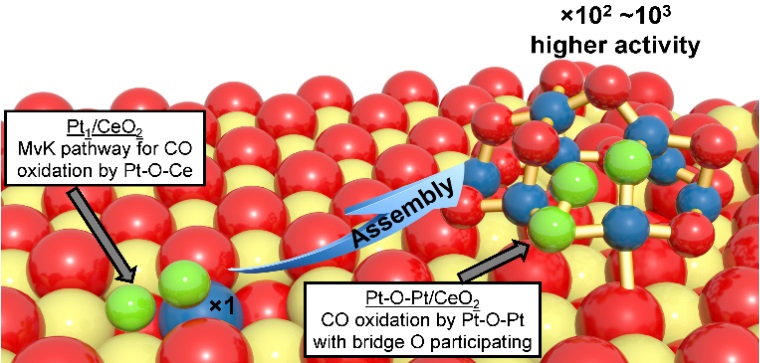
Cluster is more active than single atom catalyst for CO oxidation.
3) Electroreduction of nitrate
Nitrate (NO3⁻) pollution of aquatic ecosystems and drinking water has become a global problem and poses a threat to human health and the environment due to fertilizer overuse, wastewater discharge, and contaminant leaching from landfills. Consequently, the National Academy of Engineering recognizes the treatment of nitrate in wastewater as a grand challenge. The electrocatalytic nitrate reduction reaction (NO3RR) is a promising approach to remediate nitrate in wastewater; however, the widespread use of NO3RR is hindered because no sufficiently active, selective, and stable electrocatalyst is known. Because of the complexity of NO3RR, multiple questions remain unanswered regarding: (i) nitrate reduction electrocatalyst activity and selectivity trends across metals and alloys, (ii) the nature and abundance of the surface intermediates, and (iii) if the rate-determining step changes as a function of applied potential. Mechanistic insight into the activity and selectivity trends of NO3RR is needed to engender a framework for electrocatalyst design with superior selectivity toward benign N2 or useful NH3.
We clarified the activity and selectivity trends of transition metals for NO3RR to benign or value-added products such as N2 and NH3. Adsorbate linear scaling relations between the adsorption strengths of oxygen and nitrogen atoms with nitrate and its reduction intermediates were found on eight transition metal (i.e., Co, Cu, Rh, Pd, Pt, Ag, Au, Fe) surfaces using DFT modeling. Brønsted−Evans−Polanyi (BEP) relations were also identified for each elementary step of the nitrate reduction reaction for the considered transition metals. Nitrate reduction rates, surface species coverages, and the degree of rate control were predicted for these metals as a function of applied potential using first-principles microkinetic modeling. We compare our microkinetics simulations to our experimental NO3RR studies and reports in the literature to give mechanistic insight into nitrate reduction on metal surfaces. We predict the NO3RR activity of 28 metals and single-atom alloy electrocatalysts using adsorbate and Brønsted-Evans-Polanyi linear scaling relations with microkinetics modeling. Theoretical volcano plots and selectivity predictions, while considering metal stability, show that high activity and selectivity toward N2 or NH3 is unfeasible using pure transition metals regardless of the potential under acidic conditions. Single-atom alloys containing Fe, however, are computed to have superior selectivity to N2 or NH3 than their pure metal counterparts while maintaining activity.
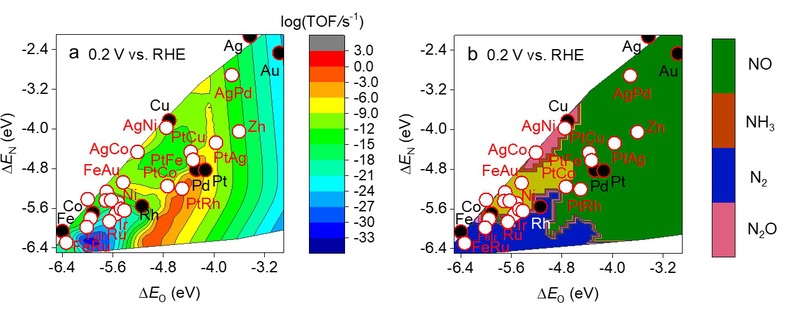
Turnover frequencies (TOF) (a) and selectivity (b) of metal and single-atom alloy catalysts for NO3RR.
