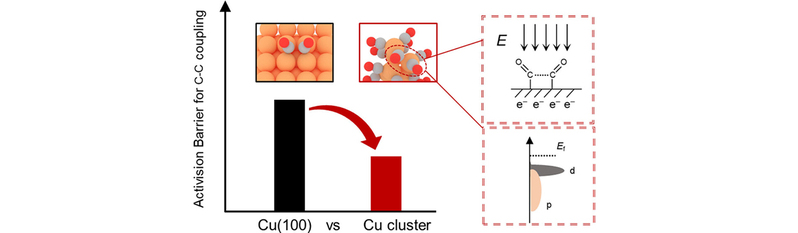
Subnanometer copper clusters supported on functional substrates have emerged as promising catalysts for electrochemical CO2 reduction (eCO2RR) to multicarbon (C2+) products. However, the mechanistic origin of their superior C–C coupling activity remains elusive. Here, we combine machine learning–accelerated grand canonical Monte Carlo sampling with grand canonical density functional theory to reveal how the electronic and structural features of the g-C3N4-supported Cu8 cluster promote CO–CO dimerization. Under increasingly negative potentials, CO adsorption is thermodynamically favored, whereas formate adsorption is suppressed, increasing both the intrinsic reactivity and the statistical likelihood of C–C bond formation. Relative to an extended Cu(100) surface, Cu8 clusters exhibit lower CO–CO coupling barriers via purely top-bound CO adsorption. This is driven by their undercoordinated Cu atoms, which incur a larger positive shift in the potential of zero charge (UPZC) and accumulate more excess electronic charge. These factors enhance Cu–OCCO orbital hybridization and stabilize the OCCO intermediate through strong electrostatic interactions induced by field–dipole coupling. Although some metastable Cu8 isomers are intrinsically active, CO-saturated global-minimum Cu8(CO)15 species dominate under operating conditions because of their high population and favorable kinetics. Our findings highlight the critical roles of the electronic structure and cluster geometry in mediating electron transfer and intermediate stabilization, yielding transferable design rules to enhance valuable-product formation across electrocatalytic platforms.